Institute of Molecular Biology
Home | About | Faculty | Calendar | Facilities | Graduate program | Contact | Apply
This page is optimized for viewing with javascript.
Marina Guenza

Professor, Chemistry and Biochemistry
Associate Member, IMB
Ph.D., University of Genoa
M.S., University of Genoa
Email
Office: 136 Klamath Hall
Office Phone: 541-346-2877
Loading profile for Marina Guenza
Research Interests
A research focus in the Guenza group is to develop computational and analytical tools for describing protein dynamics across many timescales to relate protein motion to biological function. The development of a general understanding of the relation between protein dynamics and function can provide important insights into many of their physical and biological properties. For example, kinetic processes involving protein-protein or protein-ligand interactions can depend on local conformational fluctuations. Protein stability is dominated by the thermodynamic entropy, which in turn is a function of the density of conformational states explored by the molecule and increases with increasing flexibility. Other subtle ways in which protein flexibility and dynamics are related to their biological activity include allosteric mechanisms of activation, where protein motion leads to structural fluctuations of coupled domains, which can transmit information between distant sites inside the protein. Such fluctuations can be responsible for promoting reactivity by bringing within close proximity catalytic sites that are otherwise distant. Moreover, large cooperative motions are important for protein reactivity, because they can allow substrates to access internal regions of the protein that are sterically hindered.
Because the dynamics of proteins occur over an extended range of timescales, from local bond librational motions (ps), bond re-orientation (ns), to the global tumbling and cooperative inter-domain motions (tens of nanoseconds and longer), and because of the many degrees of freedom available to the molecule, understanding the general principles of protein dynamics is a complicated problem.
Guenza’s group is developing theoretical first-principle approaches to predict protein dynamics from their structure and to understand their relation to protein function. We perform Molecular Dynamic simulations of the protein in solution and extend the timescale of motion using a Langevin approach.
An example of molecular dynamics simulation of the signal transduction protein CheY in water solvent. Only the backbone of the protein is displayed in the movie.
Our goal is to develop approaches that are general and free of adjustable parameters. To test their validity we directly compare theoretical predictions with experimental data. For example, in Figure 1 we show theoretical correlation times as a function of the protein primary sequence against those measured by NMR experiments of 1H-15N nuclear Overhauser effect, spin-lattice relaxation, and spin-spin relaxation. A peak in the spin-lattice relaxation (T1 plot) indicates that the specific residue is more mobile than the adjacent residues in the primary sequence and a faster relaxation Proteins investigated so far include the bacterial signal transduction protein CheY, the molecular motor kinesin, and the protein calmodulin.
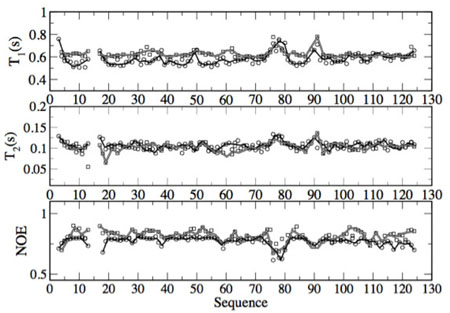 Theoretical prediction (dark lines) of NMR T1, T2 and NOE relaxation and comparison with experimental data (grey line) from Deutschman and Dahlquist. Figure from Caballero-Manrique et al. Biophys. J. 93 (12) 4128-4140 (2007).
Theoretical prediction (dark lines) of NMR T1, T2 and NOE relaxation and comparison with experimental data (grey line) from Deutschman and Dahlquist. Figure from Caballero-Manrique et al. Biophys. J. 93 (12) 4128-4140 (2007).
Recent publications
(pulled from pubmed)
Recent publications
(pulled from pubmed)
Copperman J, Guenza MG
J Phys Chem B 2015 Jul 23;119(29):9195-211
Clark AJ, McCarty J, Guenza MG
J Chem Phys 2015 Aug 14;143(6):067101
McCarty J, Clark AJ, Copperman J, Guenza MG
J Chem Phys 2014 May 28;140(20):204913
deLorimier E, Coonrod LA, Copperman J, Taber A, Reister EE, Sharma K, Todd PK, Guenza MG, Berglund JA
Nucleic Acids Res 2014 Nov 10;42(20):12768-78
Guenza MG
Phys Rev E Stat Nonlin Soft Matter Phys 2014 May;89(5):052603
Lyubimov IY, Guenza MG
J Chem Phys 2013 Mar 28;138(12):12A546
Clark AJ, McCarty J, Guenza MG
J Chem Phys 2013 Sep 28;139(12):124906
Clark AJ, McCarty J, Lyubimov IY, Guenza MG
Phys Rev Lett 2012 Oct 19;109(16):168301
Lyubimov I, Guenza MG
Phys Rev E Stat Nonlin Soft Matter Phys 2011 Sep;84(3 Pt 1):031801
Clark AJ, Guenza MG
J Chem Phys 2010 Jan 28;132(4):044902
Lyubimov IY, McCarty J, Clark A, Guenza MG
J Chem Phys 2010 Jun 14;132(22):224903
McCarty J, Guenza MG
J Chem Phys 2010 Sep 7;133(9):094904
McCarty J, Lyubimov IY, Guenza MG
J Phys Chem B 2009 Sep 3;113(35):11876-86
Caballero-Manrique E, Bray JK, Deutschman WA, Dahlquist FW, Guenza MG
Biophys J 2007 Dec 15;93(12):4128-40
Sambriski EJ, Guenza MG
Phys Rev E Stat Nonlin Soft Matter Phys 2007 Nov;76(5 Pt 1):051801
Fink MC, Adair KV, Guenza MG, Marcus AH
Biophys J 2006 Nov 1;91(9):3482-98
Sambriski EJ, Yatsenko G, Nemirovskaya MA, Guenza MG
J Chem Phys 2006 Dec 21;125(23):234902
Yatsenko G, Sambriski EJ, Guenza MG
J Chem Phys 2005 Feb 1;122(5):54907
Knowles MK, Guenza MG, Capaldi RA, Marcus AH
Proc Natl Acad Sci U S A 2002 Nov 12;99(23):14772-7